

推荐产品
联系科正
24小时业务咨询:18629290554
公司地址:陕西省西安市未央区凤城八路水晶卡芭拉11幢1单元16层11605
全国服务热线:029-86711893
业务销售咨询:18629290554 (服务专线,谢绝推销)
网址:www.xakezheng.com
邮箱:xacogent@qq.com
药品GMP认证流程及要求
责任编辑:未知
发表时间:2017-03-02


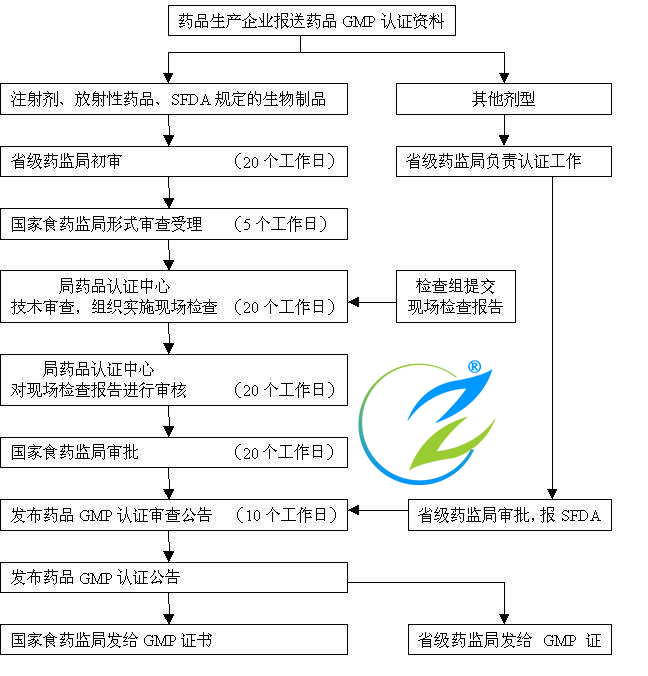
1、申报企业到省局受理大厅提交认证申请和申报材料
2、省局药品安全监管处对申报材料形式审查 (5个工作日)
3、认证中心对申报材料进行技术审查 (10个工作日)
4、认证中心制定现场检查方案(10个工作日)
5、省局审批方案 (10个工作日)
6、认证中心组织实施认证现场检查 (10个工作日)
7、认证中心对现场检查报告进行初审 (10个工作日)
8、省局对认证初审意见进行审批(10个工作日)
9、报国家局发布审查公告(10个工作日)
1.药品生产企业GMP认证
2.中药饮片GMP认证
3.原料药车间GMP认证
4.口服制剂车间GMP认证
5.片剂GMP认证
6.胶囊剂GMP认证
7.颗粒剂GMP认证
8.散剂GMP认证
9.滴丸剂GMP认证
10.栓剂GMP认证
11.注射剂GMP认证
12.放射性药品GMP认证
13.生物制品GMP认证

A 级:高风险操作区,如灌装区、放置胶塞桶和与无菌制剂直接接触的敞口包装容器的区域及无菌装配或连接操作的区域,应当用单向流操作台(罩)维持该区的环境状态。单向流系统在其工作区域必须均匀送风,风速为0.36-0.54m/s(指导值)。应当有数据证明单向流的状态并经过验证。在密闭的隔离操作器或手套箱内,可使用较低的风速。
B级:指无菌配制和灌装等高风险操作A级洁净区所处的背景区域。
C级和D级:指无菌药品生产过程中重要程度较低操作步骤的洁净区。
● 洁净度A级用于高风险作业区,如:灌装区、放胶塞区、敞口包装容器区和无菌装配区等区域。其单向流区工作区必须均匀送风,其风速为0.36 m/s ~0.54 m/s。确认A级,每个测点的采样量不得少于1 m3;洁净度为ISO4.8级,并以≥5.0 µm悬浮粒子的浓度为限度标准。采样管的长度要短,以勉≥5.0 µm的粒子沉降,影响测试结果。单向流应采用等动力采样。
● 洁净度B级用于洁净度A级区域的背景区域。静态洁净度为ISO5级。
● C级和D级用于无菌药品生产过程中工艺要求洁净较低的区域。C级静态和动态分别为ISO 7级和ISO 8级,D级静态为ISO 8级。
● 动态可采用培养基模拟灌装过程以证明达到动态洁净度级别。
日常动态监测;
● 新版GMP规定生产工作结束,作业人员离开现场经过15-20分钟自净后,洁净室的洁净度应达到“静态”标准。
● 日常动态监测项目:洁净度、温度、相对湿度、压力梯度等。
微生物的动态监测;
● 微生物的日常动态监测方法:沉降菌法、定量空气浮游菌采样法、表面取样法等。
上一篇: 百级层流配套液槽龙骨参考
下一篇: 洁净室FFU吊顶龙骨系统安装施工图解
此文关键字:药品GMP认证流程及要求







